
采用钯催化串联反应制备3-氰基吲哚。得到了“N-H”未保护的N-烷基和N-芳基3-氰基吲哚。这种合成方法的有效性通过成功合成实用化合物,如治疗性雌激素受体配体A前体得到进一步证明。机理研究表明,该串联催化利用Suzuki交叉偶联,随后碱基诱导的异恶唑碎裂,然后是醛二胺缩合。
在优化的反应条件下,多种 2-卤代苯胺2在 0.5 mol% PdCl2dppf 存在下进行串联反应,以优异的收率获得所需的“N-H”未保护的 3-氰基吲哚 3a-h . 如图1所示,值得注意的是,-NO2 和 -CF3 等官能团在反应条件下具有良好的耐受性。在吲哚的5位或6 位引入给电子或吸电子取代基对反应没有明显影响,尽管在R′位引入吸电子基团略微降低了产率(3a,3b 与 3c-g)。
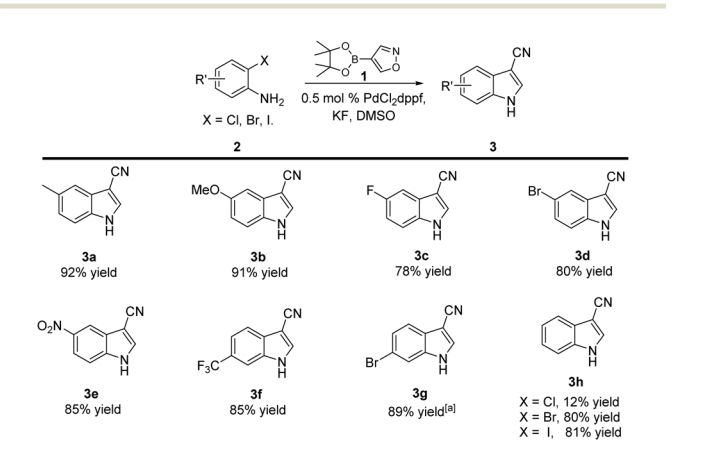
图1
根据获得的3-氰基吲哚具有游离-NH 单元 ,我们将这种串联催化的底物范围扩展到N-取代的3-氰基吲哚,包括各种 N-烷基和 N-芳基 3-氰基吲哚(图2)、N-烷基3-氰基吲哚,空间位阻的显著增加对反应活性几乎没有影响,并且在 R′位带有直链或支链烷基链的底物可以顺利反应而不会降低产率 (3i-l)。此外,在优化的反应条件下,N-芳基保护的 3-氰基吲哚也以79-94% 的收率被分离出来。N-苯基的邻位、对位或间位取代基不影响反应结果,得到的产率仍然很高(3n-x)。
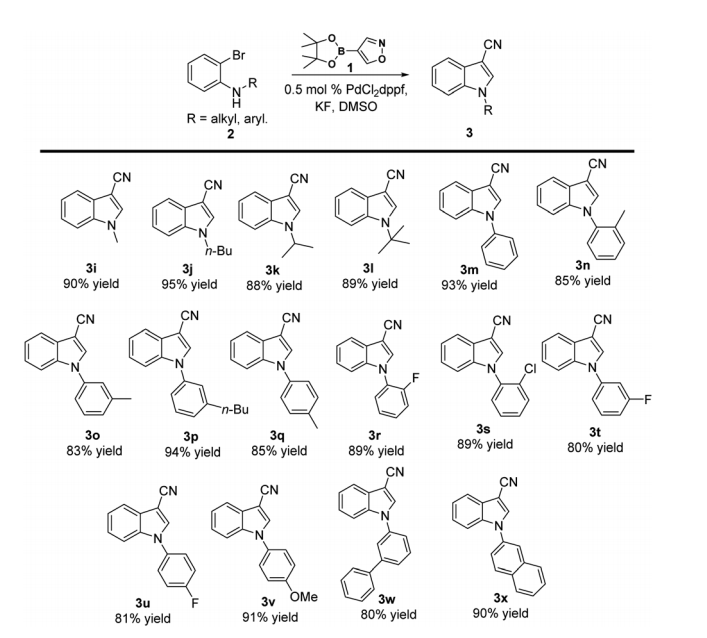
图2
为了进一步证明这种方法的合成用途,该催化方案被成功地应用于放大合成和合成转化。 在1.0 mol% PdCl2dppf的条件下,N-芳基3-氰吲哚3a和3v在克级上的分离产率超过85%(图3a)。 3v的进一步溴化使6v成为治疗性雌激素受体配体A的优良前体(图3b)。
这种串联催化的有用性也体现在3-氰基吲哚3h的实际转化中(图3c)。N-甲基类似物3i和N-苯基类似物3m可以很容易地从未保护的“N-H”游离吲哚 3h 中获得。在这项工作中,它们也可以通过串联催化直接合成(图2)。使用文献方法,3h也可以转化为其他有价值的化合物,例如羧酸7h和2-甲酰基-1H-吲哚-3-腈8h。
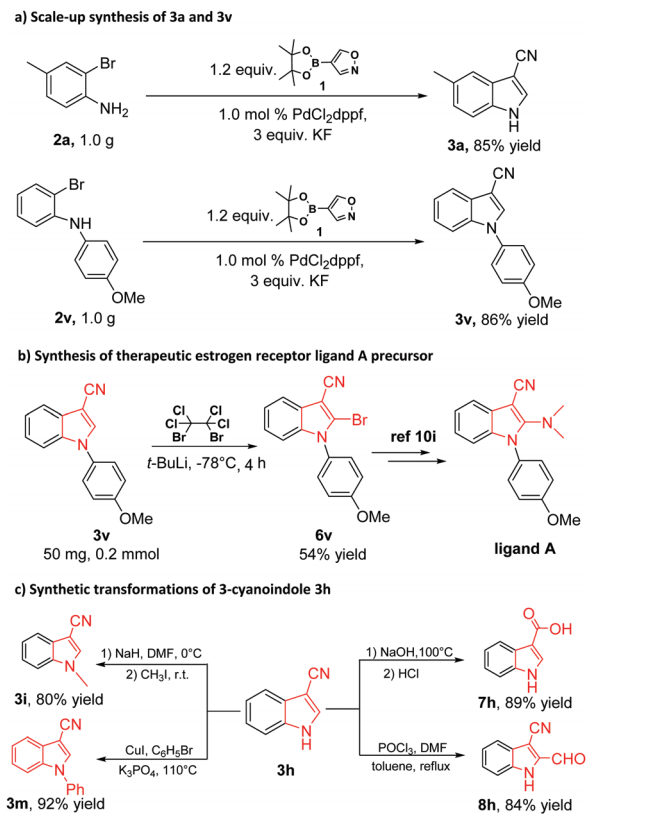
图3
对于图3形成的机理研究,我们假设主要交叉偶联产物(即异恶唑4h)的碱诱导断裂产生中间体A,而中间体A又通过醛亚胺缩合过程转化为3h。为了进一步了解串联反应过程,进行了几个对照实验。在反应条件下成功合成了交叉偶联产物异恶唑4h和4y,并与硼酸酯1反应。正如预期的那样,3h 的产率与从 2-溴苯胺(图4a)开始的标准串联反应中获得的产率相似。结果表明异恶唑4h是串联催化的中间体。至于异恶唑4y,只得到氰甲基化产物5y。
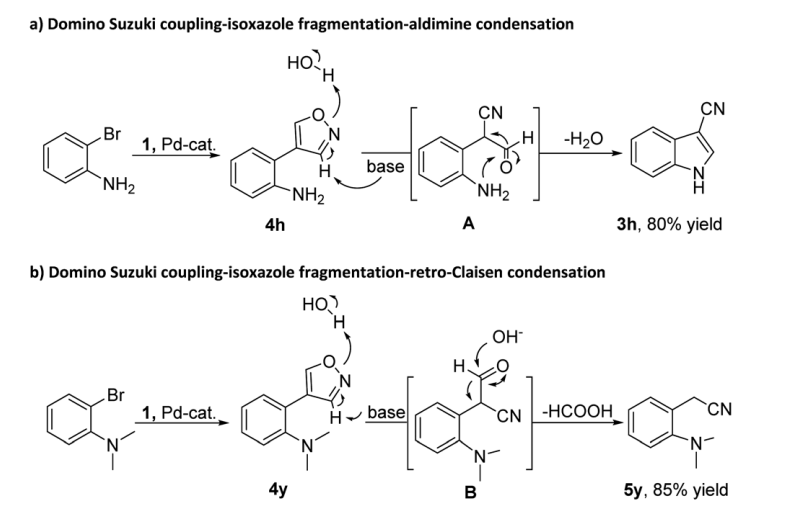
图4
总之,我们发现、优化并应用了一种直接合成 3-氰基吲哚及其衍生物的新方法。使用市售的2-卤代苯胺和异恶唑-4-硼酸频哪醇酯作为底物,以优异的产量获得了大量不受保护的氮氢化合物、N-烷基和 N-芳基 3-氰基吲哚及其类似物。串联催化利用 Suzuki 交叉偶联以及随后的碱诱导的异恶唑裂解,然后是醛亚胺缩合,从而以操作简单和可靠的方式有效地获得了用途广泛的杂环化合物。
 科研概况
科研概况  科研论文
科研论文 常用链接
常用链接